









Turinys
Cistinė fibrozė (cistinė fibrozė)
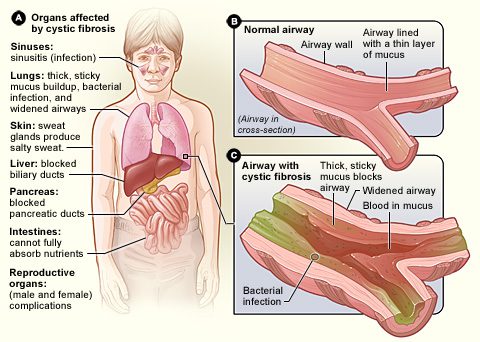
La cistinė fibrozė, čia genetinė liga dažniausiai. Pagrindinės apraiškos yra susijusios su kvėpavimo ir virškinimo traktais, tačiau gali būti pažeisti beveik visi organai. Simptomai dažnai išryškėja ankstyvoje kūdikystėje ir yra skirtingo sunkumo. Ši liga sukelia a susitraukimas gleivių, kurias išskiria sinusų, bronchų, žarnyno, kasos, kepenų ir reprodukcinės sistemos gleivinės (žr. diagramą).
Šios plaučiai dažnai yra labiausiai paveikti. The tirštos, klampios išskyros trukdo bronchams, todėl sunku kvėpuoti. Be to, plaučiuose besikaupiančios gleivės skatina mikrobų augimą. Todėl žmonėms, sergantiems cistine fibroze, yra didesnė rizika susirgti dažnomis ir galimai rimtomis kvėpavimo takų infekcijomis.
La cistinė fibrozė taip pat paliečia virškinimo sistema. Gleivės linkusios blokuoti plonus kasos latakus, neleidžiant kasos gaminamiems virškinimo fermentams patekti į žarnyną ir vykdyti savo veiklą. Kadangi maistas virškinamas tik iš dalies, ypač riebalai ir tam tikri vitaminai, atsiranda didelių trūkumų. Jie gali sukelti a augimo sulėtėjimas.
Liga taip pat turi didelių pasekmių kepenims ir reprodukciniams organams, dažnai sukelianti moterų nevaisingumą ir nevaisingumą. nevaisingumas sergančių vyrų.
A ankstesnė diagnozė ir geresnė priežiūra,gyvenimo trukmė o nukentėjusiųjų gyvenimo kokybė per pastaruosius dešimtmečius toliau gerėjo, ypač dėl to, kad pradeda atsirasti naujų gydymo būdų, skirtų genetinei anomalijai, ir vidutiniu laikotarpiu pakeis pacientų gydymą. .
Paplitimas
La cistinė fibrozė yra genetinė liga labiausiai paplitęs Prancūzijoje, nukentėjo beveik 6000 žmonių1.. Šia liga serga vienas iš 4 naujagimių. Daug rečiau pasitaiko tarp juodaodžių (000 iš 1) ir rytiečių (13 iš 000). Tai paveikia ir vyrus, ir moteris. Labiausiai nukenčia Vakarų Prancūzijos gyventojai.
La cistinė fibrozė yra genetinė liga labiausiai paplitusi liga Kanadoje. Suserga vienas iš 3 naujagimių1. Cistinė fibrozė yra šiek tiek dažnesnė Kvebekas nei likusioje Kanados dalyje: nukentėjo 3 kanadiečiai, įskaitant 500 Kvebeko gyventojų.
Priežastys
La cistinė fibrozė pirmą kartą 1936 metais aprašė Dr Guido Fanconi, Šveicarijos pediatras. Atsakingą geną, pavadintą CFTR („cistinės fibrozės transmembraninio laidumo reguliatorius“), Kanados mokslininkai nustatė tik 1989 m. Sergantiems žmonėms tai genas is nenormalus (sakome, kad jis perkeltas). Jis yra atsakingas už chloro kanalo, leidžiančio reguliuoti gleivių hidrataciją, sintezę. Atsiradus CFTR geno anomalijai, gleivės produktas yra per storas ir normaliai nenuteka. Nustatyta daugiau nei 1 skirtinga CFTR geno mutacija, susijusi su cistine fibroze23,4. Jie skirstomi į 6 klases pagal skirtingą disfunkcijos tipą2Iš šių daugelio mutacijų labiausiai paplitusi yra Delta F508 mutacija, kuri randama 81% žmonių, sergančių Prancūzijoje.
Cistinė fibrozė nėra užkrečiama liga. Žmonės, kuriems priklauso patogeninės mutacijos CFTR geno liga anksčiau ar vėliau išsivysto, bet įvairaus sunkumo.
Diagnostinis
Paprastai cistinė fibrozė diagnozuojama jau pirmaisiais gyvenimo metais, nes kvėpavimo simptomai pasirodo labai anksti. 90% atvejų liga nustatoma iki 10 metų amžiaus.
Diagnozei patvirtinti gydytojas atlieka a prakaito testas (arba prakaito testas). Iš tiesų, cistine fibroze sergančių žmonių prakaitas yra daug didesnis koncentruota druskoje (2–5 kartus daugiau nei įprastai). The genetiniai tyrimai leidžia tiksliai nustatyti CFTR geno anomalijas. Jie yra būtini svarstant tikslinę terapiją.
Prancūzijoje cistinė fibrozė sistemingai tikrinama visiems naujagimiams nuo 2002 m.5. Įrodyta, kad ankstyvas patikrinimas pagerina sergančių vaikų gyvenimo kokybę ir gyvenimo trukmę. Naujagimiai imami 3 gyvenimo dienas po tėvų sutikimo, prieš išleidžiant. motinystė. Testas nepateikia tikslios diagnozės, tačiau ją patvirtins arba paneigs specialūs papildomi tyrimai (prakaitavimo testas, genetinis tyrimas).
Kvebeke nėra sistemingas patikrinimas šios ligos. Tačiau Kanados cistinės fibrozės fondas, remiamas kelių gydytojų, jau keletą metų ragina atlikti naujagimių patikrą. Įrodyta, kad ankstyvas aptikimas pagerina sergančių vaikų gyvenimo kokybę ir gyvenimo trukmę.
Tikėtina gyvenimo trukmė
„1960“ sistemojegyvenimo trukmė vaikų, sergančių cistine fibroze, neviršijo 5 metų. Šiais laikais, remiantis naujausia statistika, vidutinis išgyvenamumo amžius yra 47 metai1. kvėpavimo takų infekcijos išlieka dažniausia mirties priežastimi.
Dažnos komplikacijos
Cistinė fibrozė yra liga, kuri palaipsniui pažeidžia plaučius, kasą ir kepenis. į medicininė stebėsena Tačiau tai padeda sumažinti komplikacijų sunkumą ir dažnį.
Šios kvėpavimo takų komplikacijos yra dažniausios, įskaitant bronchų išsiplėtimą, sukeliantį bronchitą, pneumoniją su pasikartojimais. Pasitaiko kvėpavimo takų simptomų paūmėjimo laikotarpių, kai pacientai labai „užsikimšę“, labiau kvėpuoja, krenta svoris, dažnai dėl infekcijos. Kvėpavimo sistemos pažeidimas gali būti pavojingas gyvybei.
Dėl: virškinimo sistema, užsikimšę tulžies latakai, leidžiantys tulžiui nutekėti į virškinamąjį traktą, gali sukelti kepenų cirozę. Obstrukcija ir progresuojanti sklerozė kasa, gali sukelti maistinių medžiagų malabsorbciją ir diabeto vystymąsi. Šie sutrikimai dažnai sukelia mitybos trūkumai sunkus ir lėtinis viduriavimas. Paprastai trūkumus galima ištaisyti laikantis specialios dietos. Ir atvirkščiai, gali pasireikšti didelis vidurių užkietėjimas ar net žarnyno nepraeinamumas.
Paprastai cistine fibroze sergantiems berniukams ir mergaitėms brendimas pasireiškia vėliau. Galiausiai, vaisingumas yra sumažėjo, ypač vyrams, kurie beveik visi (95 %) yra sterilūs dėl kraujagyslių obstrukcijos. Šie latakai perneša spermą iš sėklidžių į sėklines pūsleles. Moterims dėl padidėjusio makšties gleivių klampumo sulėtėja spermatozoidų judėjimas. Liga taip pat gali turėti įtakos ovuliacijos reguliarumui ir dažnumui. Vaisingumas mažėja, tačiau nėštumas vis dar visiškai įmanomas.